Pomiar masy w przemyśle farmaceutycznym
- Szczegóły
RADWAG – polski producent wag laboratoryjnych – gwarant najlepszej jakości

Wysokiej jakości wagi są grupą urządzeń, która stanowi istotny element wyposażenia firm farmaceutycznych. Produkcja leków była i jest związana z mniejszym lub większym ryzykiem popełnienia błędu. Ryzyko w tym przypadku to wprowadzenie na rynek leku szkodliwego dla pacjenta. Żeby tego uniknąć, przez lata kładziono nacisk na rozszerzenie kontroli jakości. Nie spowodowało to jednak eliminacji pomyłek. Ostatnio zwycięża koncepcja uzyskiwania wysokiej jakości leku poprzez jej zaplanowanie „Quality by Design”, a nie skontrolowanie gotowego wyrobu. W odniesieniu do jakości należy zaznaczyć, że nie może być ona oceniana przez badanie produktu. Powinna być „wbudowana w produkt” lub zagwarantowana przez projekt procesu.
W większości procesów służących do opracowania formuły leku należy dokonać pomiaru masy. Jakość tego ważenia ma dość znaczny wpływ na jakość wyniku końcowego. Stąd też wynikają kryteria akceptacji, jakie określają dla siebie firmy farmaceutyczne oraz zalecenia dla procesów ważenia, jakie przedstawia Amerykańska Farmakopea*. Takich zaleceń nie zawiera Farmakopea Europejska, stąd też te firmy, które nie podlegają wymogom amerykańskim, mają własne kryteria oceny urządzeń, jakimi są wagi laboratoryjne.
(*) Farmakopea, kodeks apteczny – urzędowy spis leków dopuszczonych w danym kraju lub na danym terenie do obrotu, oraz obwarowany tymi samymi zastrzeżeniami spis surowców służących do ręcznego sporządzania niektórych z tych leków w aptece (leków recepturowych). W Polsce oficjalna farmakopea to Farmakopea Polska wydawana przez Urząd Rejestracji Leków, Wyrobów Medycznych i Produktów Biobójczych.
Jakie rodzaje wag wykorzystuje się w farmacji?
Farmacja jako dość rozbudowana gałąź przemysłu wykorzystuje w roli przyrządów do pomiaru masy substancji szeroki wachlarz wag, w tym wagi przemysłowe i laboratoryjne.
 | Dla celów przejęć magazynowych stosowane są wagi 4-czujnikowe, które wykorzystuje się również do rozdzielania ładunków na mniejsze porcje. |
Na etapie produkcji wykorzystuje się wagi platformowe 1-czujnikowe oraz wagi precyzyjne i wagi analityczne.



W badaniach laboratoryjnych stosuje się wagi analityczne, pół-mikroanalityczne oraz mikrowagi (ultra-mikrowagi).


Nazewnictwo tych wag pokazuje poniższa tabela.
| Nazwa wagi | Rozdzielczość | Ilość cyfr dziesiętnych |
| Ultra-mikro wagi | 0,1µg | 0,0000001 |
| Mikrowagi | 1µg | 0,000001 |
| Pół mikrowagi | 0,01 mg | 0,00001 |
| Wagi analityczne | 0,1 mg | 0,0001 |
| Wagi precyzyjne | 1g ÷ 1mg | 1g ÷ 1mg |
Tabela 1. Nazewnictwo wag z uwzględnieniem rozdzielczości
Wymagania odnośnie jakości, kontroli i sprawdzania wag laboratoryjnych
Ogólne zasady dotyczące kontroli i sprawdzania wag znajdują się w Rozporządzeniu Ministra Zdrowia z dnia 1 października 2008 r. w sprawie wymagań Dobrej Praktyki Wytwarzania.
Załącznik: Szczegółowe wymagania dobrej praktyki wytwarzania.
Część 1, Rozdział 3: Pomieszczenia i urządzenia
Pkt. 3.41. Urządzenia do mierzenia, ważenia, rejestracji i kontroli powinny być kalibrowane i sprawdzane odpowiednimi metodami, w określonych odstępach czasu. Powinny być przechowywane odpowiednie zapisy dotyczące tych czynności.
Część 2, Rozdział 5: Urządzenia procesowe
Pkt. 5.3 Kalibracja
5.30 Wyposażenie do kontroli, ważenia, mierzenia, monitorowania i testowania, krytyczne dla jakości półproduktów i farmaceutycznych substancji czynnych, powinno być kalibrowane zgodnie z pisemnymi procedurami i ustalonym harmonogramem.
5.31 Kalibracja sprzętu powinna być przeprowadzana przy użyciu wzorców mających odniesienie do wzorców certyfikowanych, jeżeli takie istnieją.
5.32 Należy przechowywać zapisy dotyczące przeprowadzonych kalibracji.
5.33 Powinien być znany i możliwy do sprawdzenia aktualny status kalibracji wyposażenia krytycznego.
5.34 Urządzenia nie spełniające kryteriów kalibracji nie powinny być użytkowane.
5.35 Odchylenia od zatwierdzonych wzorców kalibracyjnych, dotyczące urządzeń krytycznych powinny być oceniane w celu stwierdzenia, czy mogły mieć wpływ na jakość półproduktów i farmaceutycznych substancji czynnych, wyprodukowanych za pomocą tych urządzeń od czasu ostatniej prawidłowej kalibracji.
Powyższe zalecenia obligują do kontroli, natomiast nie precyzują żadnych kryteriów akceptacji czy też limitów ostrzegawczych. Można stwierdzić, że firma farmaceutyczna powinna produkować wyroby zgodnie z zapisem jaki zawiera reguła w załączniku „Szczegółowe Wymagania Dobrej Praktyki Wytwarzania”, która brzmi:
„Wytwórca produktów leczniczych jest obowiązany zapewnić, żeby produkty lecznicze przez niego wytwarzane były odpowiednie do ich przewidzianego zastosowania, spełniały wymagania pozwolenia na dopuszczenie do obrotu i nie narażały pacjentów na ryzyko związane z niedostatecznym bezpieczeństwem stosowania, nieodpowiednia jakością lub zbyt niską skutecznością. Aby w sposób rzetelny osiągnąć cel jakościowy musi być szczegółowo opracowany i prawidłowo wprowadzony system Zapewnienia Jakości obejmujący Dobrą Praktykę Wytwarzania, Kontrolę Jakości oraz Zarządzanie Ryzykiem Jakości.”
Poza określeniami zawartymi w w/w rozporządzeniu stosowne zapisy odnośnie sprawdzania wag zawiera norma PN-EN ISO/IEC 17025 „Ogólne wymagania dotyczące laboratoriów badawczych i wzorcujących” pkt. 5.6.:
„Całe wyposażenie pomiarowe używane do badań . . . . , które ma znaczący wpływ na dokładność . . . . , powinno być WZORCOWANE przed oddaniem do użytkowania. Laboratorium powinno mieć ustalony PROGRAM oraz PROCEDURE wzorcowania swego wyposażenia”.
Podobny zapis znajduje się w EN ISO 9001:2008 „System Zarzadzania Jakością” pkt. 7.6.:
„... Tam gdzie niezbędne jest zapewnienie wiarygodnych wyników, wyposażenie pomiarowe należy:
a) wzorcować lub sprawdzać w wyspecyfikowanych odstępach czasu lub przed użyciem w odniesieniu do wzorców jednostek miary mających powiazanie z międzynarodowymi lub państwowymi wzorcami jednostek miary ...”
W przypadku firm pracujących zgodnie z wymogami US Pharmacopeia to stosowane zapisy zawiera dokument 21 CFR 211 US GMP Guide Drugs § 211.68:
,,Automatyczne, mechaniczne i elektroniczne wyposażenie . . . powinno być rutynowo wzorcowane, nadzorowane i sprawdzane zgodnie z zapisanym programem celem zapewnienia wymaganego działania” oraz dokument 21 CFR 211 US GMP Guide Drugs § 211.160:
Wzorcowanie urządzeń . . . zgodnie z ustanowionym zapisanym programem zawierającym szczegółowe wskazówki, harmonogramy, limity dla dokładności i precyzji . . . Urządzenia, aparatura i urządzenia rejestrujące nie posiadające określonej specyfikacji nie mogą być użytkowane”
Z powyższych dokumentów jednoznacznie wynika konieczność nadzorowania i sprawdzania urządzeń pomiarowych „wag” w sposób jednoznacznie opisany, powtarzalny celem zapewnienia odpowiedniej jakości. Ze względu na ogólny charakter zapisów kwestia jakie przyjąć kryterium przy realizacji zadania, jakim jest sprawdzanie wagi laboratoryjnej, pozostaje otwarta.
Legalizować czy wzorcować?
W pierwszej kolejności należy stwierdzić, że zgodnie z wymogami Dyrektywy 90/384/EEC rozróżnia się dwie kategorie wag, stosowanych do określenia:
a)
1. masy do transakcji handlowych;
2. masy dla obliczenia opłaty targowej, cła, podatku, premii, kary, wynagrodzenia, odszkodowania lub podobnych typów opłat;
3. masy w celu zastosowania przepisów ustawowych lub wykonawczych; ekspertyzy wydawane w postepowaniach sądowych;
4. masy w praktyce medycznej do ważenia pacjentów w celu monitorowania, diagnozowania i leczenia;
5. masy dla sporządzania lekarstw na receptę w aptece i określenia masy w analizach wykonywanych w laboratoriach medycznych i farmaceutycznych;
6. określanie ceny na podstawie masy do celów sprzedaży bezpośredniej w obrocie publicznym oraz przy paczkowaniu;
b) wszelkich innych zastosowań nie wymienionych w punkcie a.
Wagi zawarte w punkcie a) podlegają prawnej kontroli metrologicznej. W odniesieniu do nich przeprowadza się proces oceny zgodności z wymaganiami zasadniczymi dyrektywy 90/384/EEC. Efektem tego procesu jest naklejka „zielone M”.
Powyższe wymagania obligują wszystkich użytkowników przyrządów do pomiaru masy, jakimi są wagi, w przemyśle farmaceutycznym do użytkowania wag, co do których dokonano pozytywnie oceny zgodności z wymaganiami dyrektywy 90/384/EEC. Jest to wymaganie pochodzące z tzw. prawnego obszaru tzn. nadzoru państwa nad urządzeniami służącymi do pomiaru masy.
Z drugiej strony Systemy Zarzadzania Jakością oraz wytyczne GLP, GMP, FDA* wymagają od wag procedur wzorcowania, które określają rzeczywiste błędy wagi patrz pkt. 3.2.
(*) FDA, Food and Drug Administration - Agencja ds. Żywności i Leków, Amerykańska Agencja Rządowa utworzona w 1906. Wchodzi w skład departamentu zdrowia i usług społecznych i jest odpowiedzialna za kontrole żywnosci (dla ludzi i zwierząt), suplementów diety, leków (dla ludzi i weterynaryjnych), kosmetyków, urządzeń medycznych i urządzeń emitujących promieniowanie (w tym także te nie medyczne), materiałów biologicznych i produktów.
Podsumowanie
Farmacja powinna stosować wagi, które dopuszcza do użytkowania system nadzoru państwa czyli obszar prawny, choć błędy tych wag mogą być znaczne (w użytkowaniu mogą być dwukrotnie większe od błędów dopuszczalnych przy deklarowaniu zgodności). Z praktyki natomiast wiadomo, że farmacja wymaga dokładnych procesów wagowych, czyli wag z błędami dużo mniejszymi niż te wynikające z normy PN-EN 45501. Ten problem jest szczególnie widoczny przy ocenie wag pół-mikroanalitycznych, mikrowag i ultra-mikrowag.
Kryteria oceny wag
Do oceny parametrów wag w czasie użytkowania większość działów nadzoru metrologicznego przyjmuje własne kryteria. Wynikają one z oceny całości procesu wytwarzania, kontroli substancji, uwzględniając to, jak dokładnie masa próbki ma być wyznaczona. Jest to pierwsze podejście, które wymaga dobrej znajomości wymagań jakościowych procesu produkcji czy też kontroli.
Drugim podejściem może być oparcie się na uregulowaniach normy PN-EN 45501 „Zagadnienia metrologiczne wag nieautomatycznych”. Norma ta precyzuje wielkość granicznych błędów dopuszczalnych, jakie może wykazywać waga. Należy przy tym zaznaczyć, że błędy w użytkowaniu są dwukrotnie większe niż te podane w normie. Podział tych błędów pokazuje poniższa tabela.
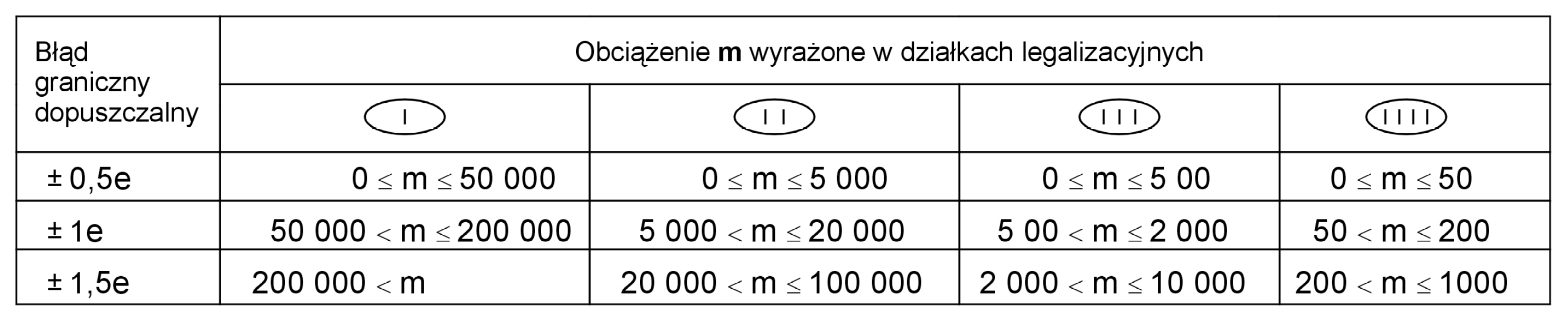
Tabela 2. Błędy graniczne dopuszczalne przy wykonywaniu procesu oceny zgodności z zasadniczymi wymaganiami dyrektywy 90/384/EEC wg normy PN-EN 45501
Praktycznie opieranie się wyłącznie na tej normie przy ocenie wag jest rozwiązaniem niepraktycznym – waga może posiadać duży błąd i jest uznawana za sprawną. Można przyjąć bardziej rygorystyczne kryteria oceny przyrządów do pomiaru masy (wag) używanych we własnym systemie. Takie założenie jest obowiązujące podczas kontroli wag w Dziale Kontroli firmy RADWAG – przyjęto, że waga jest sprawna, gdy jej błędy podczas kontroli są mniejsze niż błędów granicznych dopuszczalnych wynikających z PN-EN 45501.
ITEST £ ⅓ Mpe
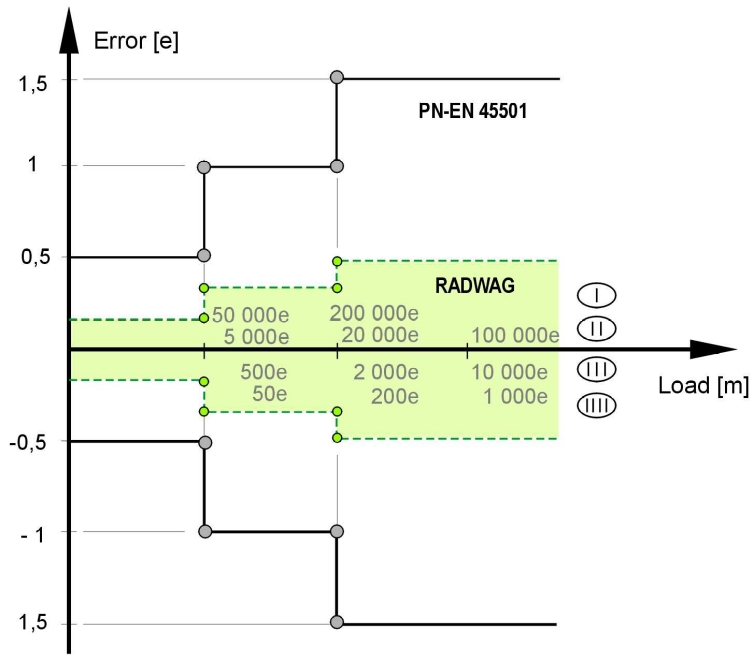
Rys.1. Graficzna interpretacja błędów granicznych dopuszczalnych wg. PN-EN 45501
Przykłady oceny wag zgodnie z wymaganiami normy pokazują poniższe tabele. Pierwsza zawiera błędy graniczne dopuszczalne dla wagi PS 600.R2. Podstawowa charakterystyka wagi:
- klasa dokładności II
- Max 600 g
- Min 20 mg
- d 1 mg
- e 10 mg
| Obciążenie m | Obciążenie | Bł. gr. dop. | Błąd gr. dop. | Rzeczywisty błąd wskazań |
| [dz. legalizac] | [g] | [dz. legalizac] | [dz. elementarna] | [dz. elementarna] |
| 0e ≤ m ≤ 5 000 e | 0 – 50 g | ± 0,5 e | ± 5 mg | ± 2 mg |
| 5 000 e < m ≤ 20 000 e | 50 – 200 g | ± 1 e | ± 10 mg | ± 3 mg |
| 20 000 e < m ≤ 51 000 e | 200 – 510 g | ± 1,5 e | ± 15 mg | ± 5 mg |
Tabela 3. Błędy graniczne dopuszczalne wg. normy PN-EN 45501 dla wagi PS 510/C/2
Druga tabela zawiera zestawienie dla mikrowagi MYA 5. Podstawowa charakterystyka tej wagi:
- klasa dokładności I
- Max 5 g
- Min 1mg
- d 1 μg
- e 1 mg
| Obciążenie m | Obciążenie | Bł. gr. dop. | Błąd gr. dop. | Rzeczywisty błąd wskazań |
| [dz. legalizac] | [g] | [dz. legalizac] | [dz. elementarna] | [dz. elementarna] |
| 0 e ≤ m ≤ 50 000 e | 0 – 5 g | ± 0,5 e | ± 0,500 mg | ± 0,003 mg |
Tabela 4. Błędy graniczne dopuszczalne wg. normy PN-EN 45501 dla mikrowagi MYA 5
Opierając własne kryteria akceptacji na normie PN-EN 45501 należy uwzględnić fakt, że dopuszczalne błędy graniczne w użytkowaniu są dwukrotnie większe. W przypadku mikrowag i ultra-mikrowag tzw. legalizacja jest dość problematyczna ze względu na drastyczną rozbieżność pomiędzy dopuszczalnymi błędami, jakie dopuszcza norma, a rzeczywistymi błędami jakie takie wagi posiadają. Oczywiście w zakresie metodyki sprawdzania norma ta daje jasne wytyczne, które mogą być powszechnie stosowane czy też korygowane do własnych potrzeb.
Trzecie podejście przy wykonywaniu oceny wag jest związane z wymaganiami jakie stawia wagom Amerykańska Farmakopea. Jest to podejście, które definiuje jaką dokładność powinna mieć waga, żeby można ją było stosować:
Jeżeli nie jest wyspecyfikowane . . . to substancja jest poprawnie zważona
(uwzględniając błąd przypadkowy i systematyczny),
gdy niepewność pomiaru urządzenia nie przekracza 0,1% odczytu.
USP, General Chapter 41 „Weights and Balances”
Niepewność pomiaru jest satysfakcjonująca
jeżeli 3-krotne standardowe odchylenie z serii co najmniej 10 powtórzeń
podzielone przez wartość średnią z serii nie przekracza 0,001.
USP, General Chapter 41 „Weights and Balances”
Takie podejście jest stosowane przez organizacje wytwarzające leki na rynek amerykański, będące częścią globalnych koncernów, które podlegają okresowym kontrolom przez FDA.
Czwarte podejście jest ukierunkowane na Analizę Ryzyka zgodnie z dokumentem EMEA* ICH Q9** „Quality Risk Management”. Dokument ten ma charakter ogólny pokazując, jak zarządzać ryzykiem. Oczywiście wymagana jest adaptacja tych założeń do wag. Określenie: co jest ryzykiem, jak je redukować i jak nim zarządzać, wymaga zdefiniowania. Obszary ryzyka będą zależne od tego, jakie ważenia są wykonywane.
*EMEA – Europejska Agencja Leków, agencja UE do koordynacji oceny i nadzoru produktów leczniczych
**ICH Q9 - Przewodnik opracowany przez grupę ekspertów dla przemysłu farmaceutycznego
Niezależnie od przyjętego kryterium oceny wag, sprawdzeniu powinny podlegać 4 parametry takie jak:
- powtarzalność,
- błąd liniowości,
- centryczność,
- zmiana czułości.
W A-BioTech oferujemy różne rodzaje wag laboratoryjnych polskiego producenta RADWAG. Wysoka jakość, duża precyzja i zaawansowanie technologiczne tych urządzeń sprawia, że stanowią one cenne wyposażenie laboratoriów farmaceutycznych. Co warto podkreślić, w naszym asortymencie znajduje się także innego rodzaju specjalistyczna aparatura laboratoryjna.